
Heat maps showing the locations of enriched SARS-CoV-2 peptides within the Spike and Nucleocapsid proteins. Adapted from Ladner et al. 2020 bioRxiv.
Highly Multiplexed Serology
During a typical year, the average adult is naturally infected with ~10 viruses, and more than 400 viral species have been shown to infect humans. Though some of these infections are asymptomatic, they all leave behind an immunological footprint. A comprehensive understanding of individuals’ antiviral immune responses (and therefore virus infection histories) has broad implications for understanding disease transmission dynamics; geographic, racial and socioeconomic health disparities; and more generally how the immune system evolves over time in response to a diverse array of external stimuli. However, our ability to decipher and utilize this rich history of pathogen encounters has been severely impacted by technological limitations – traditionally each pathogen has needed to be assayed one at a time. By capitalizing on recent technological advances, we are developing a new holistic approach to the elucidation of an individual’s viral exposure history, which we call PepSeq. We are using PepSeq technology to broadly explore antiviral reactivity and to understand how past exposures shape the immune response to novel pathogens, like SARS-CoV-2. The Ladner Lab has also built a custom software suite (PepSIRF) for analyzing highly multiplexed serological datasets. This work is being funded by NIH U24AI152172, in collaboration with the Altin Lab at TGen.

Haplotype network illustrating the replationships among Ebola virus genomes from the Western African Epidemic. Adapted from Ladner et al. 2015 Cell H&M.
Molecular Epidemiology
Infectious diseases currently account for ~20% of global human mortality, and with continued modernization, we are seeing an increase in the number of emerging disease threats. Infectious diseases also threaten the global food supply and outbreaks in important crop and livestock species often result in substantial economic losses. Fortunately, thanks to advances in sequencing technologies, genomic epidemiology is becoming a critical part of outbreak response, allowing us to track the spread and evolution of pathogens in near real-time. And, in combination with social and environmental metadata, we can use genomic analysis to understand the factors contributing to the spread of disease. Our research leverages the latest genomic technologies to study the spread of viral and bacterial pathogens in both human and wildlife populations. Notable examples include the Ebola virus disease outbreak in Western Africa and the Zika virus disease outbreak in Florida.
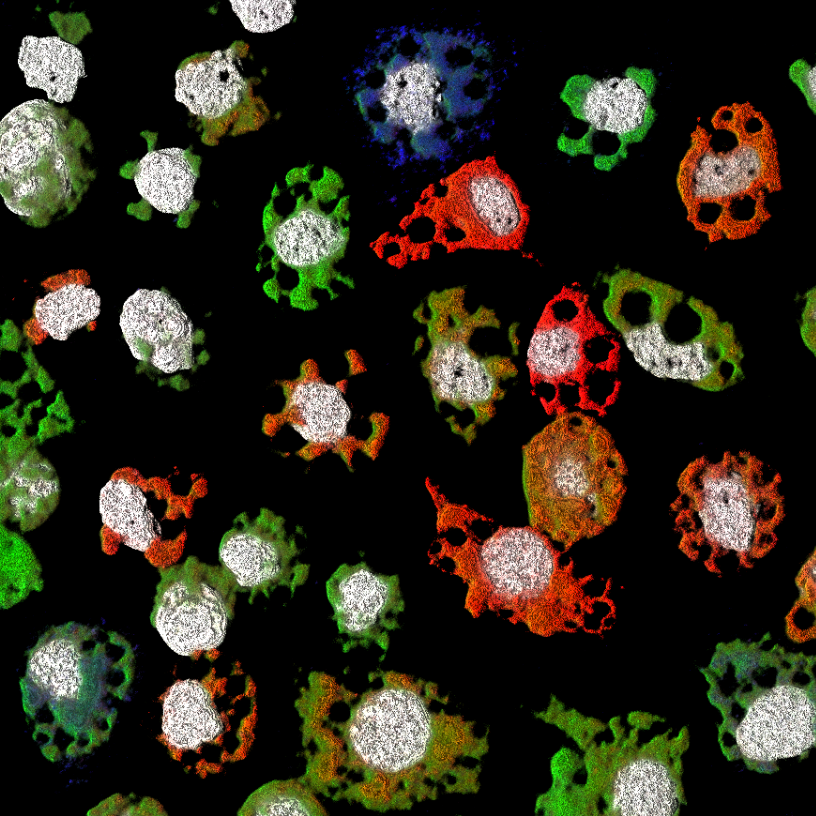
Moquito cells infected with Guaico culex virus and hybridized with RNA probes targeting different genome segments. Adapted from Ladner et al. 2016 Cell H&M.
Pathogen Discovery and Characterization
It is extremely difficult to predict what the next emerging pathogen will be, and therefore, part of our preparedness for future outbreaks must include a characterization of the extant diversity of pathogens, especially those within known reservoirs for human disease. In recent years, by utilizing high-throughput sequencing combined with metagenomic sample preparation, we have made great progress in documenting this diversity, but we still have a long way to go, and sequencing alone is insufficient for pathogen characterization. Therefore our work combines genomic data with a variety of other approaches, including proteomics, microscopy, synthetic biology and reverse genetics, to characterize novel pathogens. A recent example is Guaico culex virus (GCXV) a multicomponent virus that infects mosquitoes, and a member of the highly diverse, but only recently recognized Jingmenviruses. We are currently working to understand how these unusual viruses are naturally transmitted and the factors that impacted their evolution.

Map of defective interfering genomes within a Machupo virus population. Adapted from Golden et al. 2017 Sci. Reports.
Population Genetics and Evolution
Viruses do not exist in isolation, but rather as diverse swarms or populations, and therefore a proper understanding of their biology and pathology must include a characterization of population-level variability. In our research, we utilize emerging technologies to provide a more in-depth look into viral populations, and the ways in which they evolve in the face of changing environmetal conditions. One example is the utilization of long sequence reads from the Pacific Biosciences platform to describe linkages between the variants that arose during therapeutic treatment of Ebola virus, thus demonstrating the co-occurrence of multiple escape genotypes within a single non-human primate. I have also written custom software to characterize defective interfering genomes within viral populations. These scripts have been used to investigate persistent Ebola virus infections and to compare Machupo virus isolates with different levels of pathogenicity.